|
viernes, 28 de junio de 2019

jueves, 7 de febrero de 2019
SÍNDROME COLESTÁSICO
SÍNDROME COLESTÁSICO
 Se denomina colestasis al impedimento parcial o total del paso de bilis al duodeno, ya sea por incapacidad para su formación (en los hepatocitos) o para su flujo, causado por una gran variedad de enfermedades cuyas manifestaciones clínicas, analíticas y anatomopatológias pueden ser parecidas.
Se denomina colestasis al impedimento parcial o total del paso de bilis al duodeno, ya sea por incapacidad para su formación (en los hepatocitos) o para su flujo, causado por una gran variedad de enfermedades cuyas manifestaciones clínicas, analíticas y anatomopatológias pueden ser parecidas.
La principal característica bioquímica es el aumento sérico de los ácidos biliares, en combinación de un aumento de las enzimas marcadoras de colestasis como: fosfatasa alcalina (FA), gamaglutamiltranspeptidasa (GGT) y 5 nucleotidasa (5NT).
Esta disfunción en el metabolismo de los ácidos biliares, a menudo puede ir acompañada de alteraciones en el metabolismo de la bilirrubina (ictericia).
Los síntomas y signos de la colestasis derivan de la acumulación de sustancias que son habitualmente excretadas en la bilis, en el hígado, sangre y otros tejidos, y de la mala absorción de las grasas y vitaminas liposolubles por la disminución de ácidos biliares en el intestino delgado.
Según la localización de la alteración del flujo biliar podemos clasificar la colestasis en:
Extrahepática (obstructiva)
La obstrucción del flujo biliar puede ser consecuencia de:
- Obstrucción intraluminal
- Enfermedad obliterante de las paredes de los conductos biliares
- Compresión extrínseca
Esta forma de colestasis puede darse de modo agudo o progresivo, puede ser transitoria o persistente y presentarse como una obstrucción incompleta o completa con ictericia.
Intrahepática
Obstructiva:
Esta forma de colestasis deriva de una obstrucción permanente o temporal de los conductos biliares intrahepaticos, originado por procesos difusos que comprometen al parénquima hepático, o procesos localizados en los conductos de mayor calibre.
No obstructiva:
Los factores que causan colestasis intrahepática desencadenan disfunciones bioquímicas y/o daño en estructuras subcelulares con cambios en el metabolismo de los ácidos biliares. Además se cree que existe una predisposición genética asociada a la influencia de la raza y los hábitos (drogas, alcohol, fármacos, etc…)
Síntomas de la colestasis
- Ictericia: se ve en cuadros obstructivos con colestasis severa y prolongada.
- Purito: puede ocurrir en estadios muy tempranos, a veces siendo el primer signo de colestasis.
- Xantelasmas y xantomas: aparecen como resultado de un catabolismo deteriorado y excreción disminuida del colesterol. Son más frecuentes en colestasis crónicas.
- Malestar abdominal: el déficit de bilis en la materia fecal produce disturbios en la flora bacteriana con alteración en la digestión y absorción de las grasas, lo que produce meteorismo, distensión abdominal, náuseas y vómitos.
- Esteatorrea y diarrea: la esteatorrea biliar se caracteriza por presentar materia fecal abundante (más de 200g/día) con aumento de la excreción de grasas (más de 7gr/día). Se presenta en colestasis prolongadas.
- Malabsorción: La esteatorrea y la diarrea causan malabsorción con la consiguiente pérdida de peso y síntomas relacionados con el déficit de vitaminas liposolubles (A-D-E-K).
RECOMENDACIONES NUTRICIONALES PARA LA COLESTASIS
Debido a que en la colestasis las necesidades energéticas aumentan considerablemente, se debe administrar una alimentación densa en energía para aumentar la ingesta calórica. Para ello necesitamos darle mayor importancia a los carbohidratos y lípidos como principales fuentes de energía.
En el caso de los carbohidratos, la mejor opción es administrar polímeros de cadena corta ya que su carga osmótica no es suficientemente elevada como para causar diarrea. La cantidad que se administre depende de las necesidades estimadas en cada caso.
En cuanto a los lípidos, en la colestasis la absorción de estos disminuye considerablemente y por consiguiente la reducción de la ingesta de energía. Por lo tanto debe regularse.
Los lípidos son un complemento valiosísimo ya que tienen una densidad energética elevada, una osmolaridad baja y un contenido en AGPI esenciales.
La esteatorrea puede limitar el uso de lípidos en el apoyo nutricional. En este caso se puede proporcionar triglicéridos de cadena media ya que poseen una hidrosolubilidad elevada y son escindidos rápidamente por las lipasas. El único inconveniente es que debido a su longitud más corta de las cadenas de ácidos grasos, aportan menos contenido de energía que los triglicéridos de cadena larga.
La ingesta de proteínas debe ser en torno a 2-3gr/kg/día para compensar el catabolismo y mejorar el estado nutricional.
La mala absorción de las vitaminas liposolubles (A-D-E-K) parece ser el problema nutricional principal de la colestasis. Las recomendaciones son:
- Vitamina A
2500-500U/día. En casos de déficit aumentar hasta 25000U/día.
- Vitamina D
400-1200U/día de colecalciferol aumentando en caso de déficit hasta 1200-5000U/día.
- Vitamina K
1-3mg/día. En caso de déficit puede alimentarse por vía parenteral (100mg 1-2 veces al mes) o de un preparado hidrosoluble por vía oral.
BIBLIOGRAFÍA
Cipriani E, et al. Conversatorio clínico patológico en el Hospital Nacional Arzobispo Loayza_2009-03. Rev Med Hered [online]. 2009, vol.20, n.3 [citado 2015-11-29], pp. 169-174 . Disponible en: http://www.scielo.org.pe/scielo.php?script=sci_arttext&pid=S1018-130X2009000300009&lng=es&nrm=iso
Collares Martín, Valverde Marcelo, Fernández Isabel, Ormaechea Gabriela. Colestasis intrahepática: un desafío diagnóstico. Arch Med Int [Internet]. 2014 Mar [citado 2015 Nov 29]; 36(1): 33-38. Disponible en: http://www.scielo.edu.uy/scielo.php?script=sci_arttext&pid=S1688-423X2014000100006&lng=es.
Pérez Fernández T., López Serrano P., Tomás E., Gutiérrez Mª L., Lledó J. L., Cacho G. et al. Abordaje diagnóstico y terapéutico del síndrome colestásico. Rev. esp. enferm. dig. [Internet]. 2004 Ene [citado 2015 Nov 30]; 96(1): 60-73. Disponible en: http://scielo.isciii.es/scielo.php?script=sci_arttext&pid=S1130-01082004000100008&lng=es.
SÍNDROME DIARREICO Y ESTEATORREA
Síndrome diarreico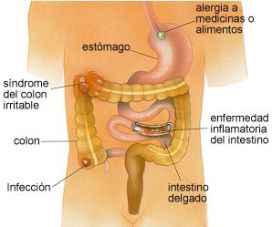
La diarrea es un desorden intestinal caracterizado por presentar deposiciones blandas y líquidas, con
un aumento en la frecuencia de
la defecación (+ de 3 veces al día) y con un peso fecal superior a 200g, además de ir acompañada de dolores abdominales.
un aumento en la frecuencia de
la defecación (+ de 3 veces al día) y con un peso fecal superior a 200g, además de ir acompañada de dolores abdominales.
Tipos de diarrea
La clasificación de diarrea desde un punto de vista fisiopatológico es:
• Diarrea osmótica
• Secretora
• Inflamatoria.
• Secretora
• Inflamatoria.
Según la duración de la diarrea, puede considerarse:
-Aguda (menos de 4 semanas):
Causas: La causa principal es infecciosa (viral, bacteriana, parasitaria), también secundaria al tratamiento con antibióticos u otros fármacos.
Complicaciones: la principal es la deshidratación y los trastornos hidroelectrolíticos.
Recomendaciones nutricionales:
• Se busca reponer agua y electrolitos con soluciones de rehidratación adecuadas
• El ayuno solo se justifica mientras exista una contraindicación médica para la administración de alimentos por la vía oral.
• El suministro nutricional oral debe iniciarse lo más tempranamente posible, debido a que los alimentos tienen efecto trófico sobre la mucosa intestinal.
• En cuanto a la consistencia la dieta debe seguir una progresión adecuada a la tolerancia del paciente: iniciando con dieta líquida, papilla, blanda y hasta llegar a la dieta normal.
• Se recomienda la ingesta de alimentos con prebióticos como pectina e inulina, así como el uso de probióticos (levaduras y bacterias) para controlar la diarrea al estimular la recuperación de la flora bacteriana normal.
• Hay que evitar los lácteos, los alimentos ricos en fibra como vegetales crudos, frutas con cáscara, leguminosas. También hay que evitar la fructosa, sacarosa, cafeína y las bebidas alcohólicas fermentadas.
Complicaciones: la principal es la deshidratación y los trastornos hidroelectrolíticos.
Recomendaciones nutricionales:
• Se busca reponer agua y electrolitos con soluciones de rehidratación adecuadas
• El ayuno solo se justifica mientras exista una contraindicación médica para la administración de alimentos por la vía oral.
• El suministro nutricional oral debe iniciarse lo más tempranamente posible, debido a que los alimentos tienen efecto trófico sobre la mucosa intestinal.
• En cuanto a la consistencia la dieta debe seguir una progresión adecuada a la tolerancia del paciente: iniciando con dieta líquida, papilla, blanda y hasta llegar a la dieta normal.
• Se recomienda la ingesta de alimentos con prebióticos como pectina e inulina, así como el uso de probióticos (levaduras y bacterias) para controlar la diarrea al estimular la recuperación de la flora bacteriana normal.
• Hay que evitar los lácteos, los alimentos ricos en fibra como vegetales crudos, frutas con cáscara, leguminosas. También hay que evitar la fructosa, sacarosa, cafeína y las bebidas alcohólicas fermentadas.
-Diarrea crónica (+ de 4 semanas):
Causas: Puede ser infecciosa (viral, bacteriana, parasitaria), por enfermedad inflamatoria intestinal, síndromes de malabsorción, cirugía gastrointestinal, trastornos funcionales crónicos (síndrome de intestino irritable)
Complicaciones: deshidratación, trastornos hidroelectrolíticos y a largo plazo deterioro en el estado nutricional con pérdida de peso y malnutrición.
Recomendaciones nutricionales:
• Tomas de alimentos en pequeños volúmenes pero más frecuentes
• Servir los alimentos a temperatura ambiente evitando las temperaturas extremas
• Evitar los irritantes: purinas, café, grasas y frituras, condimentos, alcohol
• Dieta blanda: baja en fibra y libre de residuo
• Controlar consumo de sodio
• Evitar los alimentos identificados que generen intolerancia.
Complicaciones: deshidratación, trastornos hidroelectrolíticos y a largo plazo deterioro en el estado nutricional con pérdida de peso y malnutrición.
Recomendaciones nutricionales:
• Tomas de alimentos en pequeños volúmenes pero más frecuentes
• Servir los alimentos a temperatura ambiente evitando las temperaturas extremas
• Evitar los irritantes: purinas, café, grasas y frituras, condimentos, alcohol
• Dieta blanda: baja en fibra y libre de residuo
• Controlar consumo de sodio
• Evitar los alimentos identificados que generen intolerancia.
ESTEATORREA (diarrea por malabsorción)
Caracterizada por presentar heces con alto contenido en grasa, pérdida de peso, distensión abdominal y manifestaciones propias de déficit de vitaminas y nutrientes esenciales.
El defecto de malabsorción puede localizarse en 3 niveles:
-Luz intestinal (mala digestión)
Síntomas: dolor abdominal en caso de pancreatitis, con mayor cantidad de grasa en las heces. Los pacientes no suelen presentar gases debido a que la amilasa salival y gástrica completa parcialmente la digestión y posterior absorción de los carbohidratos)
Causas: La mala absorción a este nivel puede ser originada por una disminución de la carga de ácidos biliares en la luz intestinal, como sucede en la cirrosis hepática y la insuficiencia pancreática exocrina.
Causas: La mala absorción a este nivel puede ser originada por una disminución de la carga de ácidos biliares en la luz intestinal, como sucede en la cirrosis hepática y la insuficiencia pancreática exocrina.
-En la mucosa
Síntomas: anorexia, letargia y malestar, las heces flotan en el agua debido a la presencia de gas y grasa, las proteínas séricas son bajas debido a pérdida de las mismas desde la pared intestinal, y suele haber anemia por deficiencia de folato y hierro.
Las causas que afectan directa o indirectamente a las funciones que desarrolla la mucosa intestinal son:
• Drogas: provocan daño epitelial.
• Infecciones intestinales
• Enteropatías autoinmunes
• Enfermedades del sistema inmune
• Sprue celíaco: enfermedad clásica que describe el síndrome de malabsorción
• Dermatitis herpetiforme
• Enfermedad de Whipple
• Abetalipoproteniemia
Las causas que afectan directa o indirectamente a las funciones que desarrolla la mucosa intestinal son:
• Drogas: provocan daño epitelial.
• Infecciones intestinales
• Enteropatías autoinmunes
• Enfermedades del sistema inmune
• Sprue celíaco: enfermedad clásica que describe el síndrome de malabsorción
• Dermatitis herpetiforme
• Enfermedad de Whipple
• Abetalipoproteniemia
-Fuera del intestino (submucoso)
En este nivel la causa de la malabsorción es la obstrucción linfática que impide el transporte de nutrientes, específicamente grasas y aminoácidos, que son transportados por esta vía.
-Causas mixtas (combinación de las anteriores)
Dentro de este nivel hay varias causas como el sobrecrecimiento bacteriano (SCB), que producen desconjugación de las sales biliares con pobre formación de micelios, malabsorción de grasas, inflamación epitelial, trastorno secretorio y alteración de la motilidad. Otras causas de esteatorrea mixta son el síndrome de intestino corto en donde hay una disminución de la superficie absortiva, y algunas enfermedades metabólicas como la tirotoxicosis, la insuficiencia adrenal y la desnutrición proteico-calórica.
Recomendaciones nutricionales para la esteatorrea:
Es esencial recuperar lo antes posible una situación nutricional normal, por lo tanto hay que tener un conocimiento previo de los déficits presentes. La mayoría de los déficits de oligoelementos, minerales y vitaminas pueden tratarse con los complejos vitamínicos disponibles en el mercado.
Pueden ser muy útiles los diversos preparados de nutrición enteral ya que son fáciles de tomar para pacientes en condiciones precarias, además no contienen gluten ni lactosa, lo que es importante en circunstancias específicas. También pueden tener valor terapéutico intrínseco en algunas circunstancias, como la enfermedad de Crohn. Por otro lado proporcionan proteínas en forma de péptidos simples facilitando así la asimilación, en algunos procesos. Y por último contienen oligoelementos y vitaminas, lo que disminuye la necesidad de utilizar otros suplementos.
En cuanto a las grasas, hay que suministrar triglicéridos de cadena media que no requieren de sales biliares para su emulsificación y absorción.
Recomendaciones nutricionales para la esteatorrea:
Es esencial recuperar lo antes posible una situación nutricional normal, por lo tanto hay que tener un conocimiento previo de los déficits presentes. La mayoría de los déficits de oligoelementos, minerales y vitaminas pueden tratarse con los complejos vitamínicos disponibles en el mercado.
Pueden ser muy útiles los diversos preparados de nutrición enteral ya que son fáciles de tomar para pacientes en condiciones precarias, además no contienen gluten ni lactosa, lo que es importante en circunstancias específicas. También pueden tener valor terapéutico intrínseco en algunas circunstancias, como la enfermedad de Crohn. Por otro lado proporcionan proteínas en forma de péptidos simples facilitando así la asimilación, en algunos procesos. Y por último contienen oligoelementos y vitaminas, lo que disminuye la necesidad de utilizar otros suplementos.
En cuanto a las grasas, hay que suministrar triglicéridos de cadena media que no requieren de sales biliares para su emulsificación y absorción.
BIBLIOGRAFÍA
Pineda L, Otero W, Arbeláez V, et al. Diarrea crónica. Diagnóstico y evaluación clínica. Revista Colombiana de Gastroenterología. [Internet]. 2 004; vol.19 no.2. Disponible en: http://www.scielo.org.co/scielo.php?script=sci_arttext&pid=S0120-99572004000200009
Sweetser S. Evaluating the Patient With Diarrhea: A Case-Based Approach. Mayo Clinic Proceedings. [Internet]. 2012; 87(6): 596-602. Disponible en http://www.ncbi.nlm.nih.gov/pmc/articles/PMC3538472/
FALLO DE LA SECRECIÓN INTESTINAL. INTOLERANCIA A LA LACTOSA
FALLO DE LA SECRECIÓN INTESTINAL. INTOLERANCIA A LA LACTOSA
La intolerancia a la la lactosa es bastante frecuente en la población occidental. Así, en Europa se estima en un 15-80% de la población adulta, dependiendo del origen étnico. Se debe a la disminución de actividad de una disacaridasa intestinal (lactasa), que puede deberse a su vez, a causas congénitas o ser secundaria a enfermedades del intestino delgado(enteritis virales, enfermedad de Crohn, etc…).


Es mucho más frecuente la deficiencia de lactasa intestinal que su ausencia, por lo que estos pacientes suelen tolerar pequeñas cantidades de alimentos que contengan lactosa, siendo el nivel de tolerancia individual muy variable.

La lactosa no digerida avanza a lo largo del intestino, retiene agua por efecto osmótico y es sometida a procesos de fermentación por la flora bacteriana del colon, lo que aumenta la producción de ácidos grasos, dióxido de carbono e hidrógeno, originándose de esta forma dolor cólico abdominal, meteorismo y diarrea, que aparecen entre minutos y horas después de la ingesta de lactosa.
En lo que respecta al tratamiento nutricional, es conveniente conocer el nivel individual de tolerancia, adicionando a una dieta sin lactosa pequeñas cantidades de ésta. Una vez establecido el grado de tolerancia, se puede planificar más facilmente la alimentación total.
En general se tolera mejor si se acompaña de otros alimentos, por lo que se debe aconsejar a estos pacientes que no consuman leche sola. 
La adicción a sustancias que actúen sobre vaciamiento gástrico, en el sentido de enlentecerlo, como los alimentos sólidos o la fibra, así como la sustitución de la leche por productos lácteos de mayor viscosidad, ha demostrado ser un método efectivo que mejora la tolerancia a la lactosa.
El yogur y la leche fermentada pueden mejorar la digestión de la lactosa y aliviar los síntomas gastrointestinales en las personas con malabsorción de la lactosa. Estos alimentos no sólo proporcionan menores cantidades de lactosa que la leche, sino que, además, contribuyen a retrasar el transito intestinal, lo que da más tiempo de acción a la lactasa intestinal, por lo que suelen ser mejor tolerados en la mayoría de los casos.
Además el uso de suplementos de calcio puede ser recomendable en pacientes con intolerancia a la lactosa, por ser frecuente en ellos la restricción en el consumo de lacteos.
bibliografia:
R.M Ortega, A. M. Requejo. Manual de Nutrición Clínica. Ed. Panamericana.2ª edición. 2015
Suscribirse a:
Entradas (Atom)